
歐盟仿制藥開發(fā)注冊(cè)的一般流程
總體來說歐盟的仿制藥申報(bào)的流程和注意事項(xiàng)比美國要更加零散,主要因?yàn)闅W盟由28個(gè)國家組成,藥政法規(guī)體系和美國有較大不同;另外,美國的仿制藥很多質(zhì)量要求和政策比較細(xì)化清晰,而歐盟有些法規(guī)特別是涉及到技術(shù)細(xì)節(jié)的法規(guī)不如美國明確。所以歐盟仿制藥申報(bào),操作起來并不是很方便。
本次主題涉及到歐盟仿制藥申報(bào)的多個(gè)方面,主要包括:
-
1.歐盟概況;
-
2.歐盟藥政監(jiān)管機(jī)構(gòu)概況,特別要提及第三方機(jī)構(gòu);
-
3.歐盟藥物注冊(cè)的分類;
-
4.歐盟藥品評(píng)審的程序和各個(gè)程序的特點(diǎn);
-
5.歐盟仿制藥專屬期保護(hù);
-
6.歐盟的評(píng)審費(fèi);
-
7.歐盟的原料藥的注冊(cè),及如何配合制劑申報(bào);
-
8.歐盟的QP制度就是質(zhì)量受權(quán)人制度,這個(gè)和美國有較大的不同;
-
9.歐盟的藥物警戒,這個(gè)和美國有相當(dāng)大的不同,至少在藥品注冊(cè)申請(qǐng)階段;
-
10.典型的仿制藥申報(bào)的流程;
-
11.歐盟申報(bào)資料和美國申報(bào)資料的不同。
1、歐盟概況及簡介
因?yàn)槭俏⑿湃赫Z音交流的方式,不可能講的很詳細(xì),所以挑特別重點(diǎn)的。在地理上,歐洲有五十多個(gè)國家;我們常說的歐盟,就是在我們藥品這個(gè)研發(fā)領(lǐng)域里面,只有28個(gè)國家。大家很容易判斷:英國,法國,德國肯定是屬于歐盟。注意了,有一些我們傳統(tǒng)意義上分得不太清楚的國家,其實(shí)都不屬于歐盟,比如說土耳其,敘利亞,俄羅斯,烏克蘭,他們都不屬于歐盟。相反,與此類似的還有希臘,這幾年被社會(huì)動(dòng)蕩和經(jīng)濟(jì)發(fā)展折騰得很難受,又確實(shí)是屬于歐盟。
既然談到歐盟的28國,自然大家就會(huì)想到,這幾年鬧得影響大的就是脫歐,英國傳統(tǒng)意義上肯定屬于歐盟,目前的歐盟EMA的總部也是設(shè)在英國。受英國脫歐的影響,我們藥品研發(fā)的影響就在于,參比制劑購買時(shí),如果國內(nèi)企業(yè)要買歐盟的產(chǎn)品,就不太建議買英國的。英國脫歐以后對(duì)藥政的影響,還存在不確定性。
歐盟28個(gè)國家是哪28個(gè)國家?這個(gè)網(wǎng)上有很詳細(xì)的列單。
http://www.hma.eu/nationalcontacts_hum.html,這個(gè)是歐盟的HMA網(wǎng)站,這是一個(gè)人用藥品的一個(gè)協(xié)調(diào)機(jī)構(gòu)。這個(gè)網(wǎng)址給出了歐盟的28各國成員的列表,從這個(gè)列表上我們能看到有31個(gè)國家,這就又涉及到歐盟EU和歐盟經(jīng)濟(jì)區(qū)EEA的概念了。歐盟經(jīng)濟(jì)區(qū),它是指歐盟28個(gè)國家,另外再加上冰島、挪威、和列支敦士登。這三個(gè)國家的藥政管理是和歐盟是相互認(rèn)可的。
當(dāng)然本次交流,偏向于實(shí)戰(zhàn)。這些法規(guī)和政治上的界定就不多詳述了。大家關(guān)心的一般都是比較出名的一些國家,比如德國、英國、法國、意大利、西班牙類似這樣的國家。而羅馬尼亞、保加利亞這樣的國家,它雖然屬于歐盟,但是中國醫(yī)藥行業(yè)的一般都不會(huì)去關(guān)注。
2、藥政監(jiān)管三個(gè)機(jī)構(gòu)
歐盟藥政監(jiān)管的三個(gè)比較重要的機(jī)構(gòu),第一個(gè)是EMA,想必大家都非常熟悉,不用介紹。第二個(gè)是EDQM,這個(gè)也大家也很熟悉,簡單說它的職能的就是負(fù)責(zé)歐洲藥典EP和阿歐州藥典的對(duì)照品等相關(guān)業(yè)務(wù)和監(jiān)管。第三個(gè)機(jī)構(gòu)就是我們最想和大家詳細(xì)介紹的就是HMA。HMA網(wǎng)站的地址是http://www.hma.eu/
HMA全稱是是Heads of MedicinesAgencies。他的下屬在他的下面有一個(gè)CMDh是一個(gè)人用藥品的協(xié)調(diào)合作組織。大家就可以把它理解成歐盟各個(gè)國家之間的一個(gè)協(xié)調(diào)機(jī)構(gòu)。
在HMA網(wǎng)站上有一個(gè)非常重要的功能,就是關(guān)于歐盟的仿制藥信息查詢的數(shù)據(jù)庫。歐盟的仿制藥信息不太好查,雖然部分國家的藥政機(jī)構(gòu),有自己的藥政網(wǎng)站和藥品清單或者數(shù)據(jù)庫,但是HMA是一個(gè)匯總的地方。但是,根據(jù)我們的經(jīng)驗(yàn),該網(wǎng)站對(duì)于藥品或者是仿制藥藥品的收錄,也不是很全面,有漏掉的情況。不管怎么說,這是一個(gè)比較重要的一個(gè)信息匯總。
http://mri.cts-mrp.eu/Human/Product/AdvancedSearch
這個(gè)網(wǎng)址還有個(gè)作用,大家知道美國的仿制藥的評(píng)審報(bào)告是很少公開的,即使公開偶爾公開也只有極小一部分內(nèi)容。而歐盟,在上面這個(gè)網(wǎng)站里面除了能夠檢索到仿制藥的批準(zhǔn)情況。還有一部分產(chǎn)品也能檢索到PAR,Public Assessment Report就是仿制藥的評(píng)審報(bào)告,這對(duì)反仿制藥企業(yè)是非常有用的。可以去參考別人的技術(shù)內(nèi)容,比如別人選擇的參比制劑是啥。但是也不用指望從這個(gè)報(bào)告發(fā)現(xiàn)詳細(xì)的技術(shù)細(xì)節(jié),畢竟只是一個(gè)公開的評(píng)審報(bào)告。
談到PAR,除了HMA網(wǎng)站可以檢索得到,有一些歐盟境內(nèi)的國家制劑的藥政網(wǎng)站也能檢索到一些。HMA只是一個(gè)選擇性的匯總,注意并不是說檢索了HMA,歐盟信息就全了,有些PAR只在這個(gè)國家自己的官網(wǎng)上公開,比如英國的部分PAR,HMA上收錄的并不是很全。所以,如果要檢索全面,HMA網(wǎng)站要看,各個(gè)國家的監(jiān)管機(jī)構(gòu)網(wǎng)站也要看。
3、歐盟藥物申請(qǐng)注冊(cè)分類簡介
在美國藥品申報(bào)大的分類分為505(b)1, 505(b)2, 505j三類,在歐盟也有這樣的分類。他都是根據(jù)法規(guī)的條款序號(hào)來給的這個(gè)簡稱,在歐盟也有類似的定義,具體如下:
-
Article 8(3)約等于505(b)1新藥
-
Article 10(1)約等于505(j)仿制藥
-
Article 10(3)約等于505(b)2改良型
這個(gè)只是法規(guī)上的定義,大家做仿制藥的都理解這三類的含義,就不再贅述了,具體到法規(guī)條款,網(wǎng)上也很容易查到。
4、歐盟藥品評(píng)審程序簡介及各程序的區(qū)別
歐盟藥品評(píng)審程序以及各程序的區(qū)別,歐盟的程序是比較復(fù)雜的,我們可以把它分為四個(gè)CP、MRP、DCP、NP。
第一類就CP集中程序,他是指這個(gè)藥品的申請(qǐng)上市是向EMA提交的。我們知道EMA是歐盟的一個(gè)藥政監(jiān)管的最高級(jí)別,可以類似的把它理解成美國的FDA,他是歐盟最大的監(jiān)管機(jī)構(gòu),那么如果向EMA提交新藥上市申請(qǐng),也可以包括仿制藥,如果最終獲批,就可以在歐盟所有國家上市,這個(gè)還是非常有誘惑力的。
但實(shí)際操作當(dāng)中并沒有很多的企業(yè),特別是仿制藥企業(yè)去選擇CP程序申請(qǐng)仿制藥上市。當(dāng)然,也不是說沒有,只是相對(duì)來說比較少。有一些大的仿制藥廠家,為了拿下整個(gè)歐盟市場(chǎng),認(rèn)為藥品非常有潛力,他也會(huì)走系CP程序來申請(qǐng)。
藥物上市申請(qǐng)走CP程序向EMA提交申請(qǐng)。好處就是一旦批準(zhǔn)在全盟都可以賣。但是也有壞處,一般來說EMA的評(píng)審相對(duì)來說比較嚴(yán)格,比單個(gè)國家評(píng)審要嚴(yán)格一些,EMA的專家多;另外,EMA評(píng)審業(yè)務(wù)的費(fèi)用也比較貴,一般比單一國貴。所以大部分仿制藥企業(yè),特別是中國的仿制藥企業(yè),基本上不可能走CP程序。
既然不走CP,從實(shí)戰(zhàn)的角度,該選哪一種程序呢?對(duì)于大部分的中國企業(yè)來說,選擇的是DCP,或者說絕大部分的歐盟企業(yè),他們也是選擇DCP。DCP程序就是指去中心化程序。在介紹DCP和MRP的時(shí)候,就必然離不開RMS和CMS的概念。
RMS是指參比成員國,CMS是指相關(guān)成員國。在中國的習(xí)慣翻譯,會(huì)把RMS翻譯成主審國,CMS副審國。一個(gè)仿制藥在選擇進(jìn)入歐盟的時(shí)候呢,選擇DCP程序,你需要把你的資料同時(shí)提交給主審國和副審國。主審國一般只有一個(gè)國家,副審國可以有多個(gè)國家。中國的藥企,在歐盟申報(bào)仿制藥,一般主審國選一個(gè)國家,副審國選擇也比較少,一般選擇2-5個(gè)。
為什么仿制藥,特別是我們從實(shí)戰(zhàn)的角度出發(fā),中國的企業(yè)大多選擇DCP?當(dāng)你的仿制藥還從來沒有在歐盟獲批上市,是第一次申報(bào),在DCP和MRP之間,只能選擇DCP程序,先在幾個(gè)國家去上市。
MRP指互認(rèn)程序,他指的是指你的藥品已經(jīng)在某少數(shù)幾個(gè)國家上市,以后又想擴(kuò)展更多的國家上市,這時(shí)候就要走M(jìn)RP程序。比如,第一次申請(qǐng)的選擇德國主審國,意大利和英國是副審國,合計(jì)三個(gè)國家上市后。又發(fā)現(xiàn)這個(gè)藥在西班牙也有市場(chǎng),想在西班牙也上市。這時(shí)候你就要走M(jìn)RP程序,向西班牙提交就可以了。
當(dāng)然,你也可以同時(shí)走多個(gè)DCP,比如說,德國主審,英國和意大利是副審,算一個(gè)DCP,這些都是西歐的發(fā)達(dá)國家。隨后又發(fā)現(xiàn),東歐的欠發(fā)達(dá)國家也有市場(chǎng),可以在東歐也再報(bào)另一個(gè)DCP,比如希臘作為主審國,匈牙利作為副審國。
最后一個(gè)最簡單的NP,是指單一國程序,中國藥企很少采用,不再詳細(xì)介紹。
從事歐盟藥政的人,對(duì)于CP,DCP,MRP,NP的使用范圍應(yīng)該是要相當(dāng)熟悉的。因?yàn)楸敬谓涣鞯闹黝}是仿照藥申報(bào),就不展開講了。特別是像CP程序,有很多類型的申請(qǐng),是強(qiáng)制要走CP程序的。簡單概括,一般來說原研、還有在生物制品、抗艾滋病藥、孤兒藥等等,它大多要走CP程序,這類藥品一旦上市,就普及整個(gè)歐盟。
在談這四個(gè)程序的時(shí)候,我們剛才也介紹了主審國和副審國的概念。后面還會(huì)介紹主審國和這個(gè)副審國選擇時(shí)的一般考慮。
5、歐盟仿制藥專屬期保護(hù)
再聊聊歐盟仿制藥的專屬期保護(hù)。美國想必大家都非常熟悉的,就不過多介紹。這里簡單描述歐盟的專屬期保護(hù),簡稱為8+2+1模式。8是指原研藥被批準(zhǔn)后8年仿制藥才可以提交申報(bào)。2是指2年后才能獲批。1是指如果原研藥持有人在上市8年之內(nèi)有新增適應(yīng)癥,這種情況在8+2+1年之內(nèi)是不可能被批準(zhǔn)的。
歐盟的專屬期保護(hù)和專利保護(hù)是完全獨(dú)立的兩條線,他和美國不一樣,美國的Orange Book里面會(huì)把專利列出來,因?yàn)橄雸?bào)美國ANDA的話。你肯定要針對(duì)這些專利,寫一份專利聲明,聲明專利是P1還是P2或P3或P4。而在歐盟,藥政當(dāng)局是不管你專利什么時(shí)候到期。專屬期保護(hù)到期了你就可以申報(bào),10年或者11年到了就可以批準(zhǔn)上市。
但歐盟也是有專利保護(hù)的,但不屬于歐盟藥政官方管。歐盟藥政官方只管專屬期,不管專利保護(hù)。談到歐盟的專利,必然要談到專利補(bǔ)償,我們知道美國有專利補(bǔ)償,當(dāng)然歐盟也有。一般專利是二十年專利保護(hù)期,歐盟有專利補(bǔ)償也有叫專利延長,歐盟叫SPC,這個(gè)和美國的叫法上不一樣。歐盟的SPC最多也是五年。
可以有這么一個(gè)概念,就是專屬期保護(hù)最長是11年。專利保護(hù)它是平行的,另外一條線。過了11年,你可以不管專利情況提交申報(bào),官方也可以批,但是你不敢上市,因?yàn)橛性袑@膯栴}。我們現(xiàn)在看到很多仿制藥已經(jīng)在歐盟批準(zhǔn)了,但他其實(shí)不敢上市,因?yàn)榱硗膺€有專利保護(hù)。
另外,也有很重要的一個(gè)原因,歐盟的知識(shí)產(chǎn)權(quán)保護(hù),相對(duì)來說比美國歷史要悠久,體系也非常嚴(yán)格。為什么很多仿制藥的廠家在美國發(fā)起一輪又一輪的專利挑戰(zhàn),而歐盟相對(duì)來說沒有那么活躍呢?另外一個(gè)原因就是,在歐盟假設(shè)你報(bào)主審國1個(gè)國家副審國4個(gè)國家,共5個(gè)國家上市。你是因?yàn)閷@麊栴}提前上市你需要分別在5個(gè)國家去打官司。程序上也很麻煩。
6、仿制藥繳費(fèi)
大家知道美國的仿制藥非常貴,申報(bào)費(fèi)用大概就要17萬美元左右。歐盟的仿制藥,按照DCP程序來走,其實(shí)是比較便宜,甚至可以說非常便宜。但是這個(gè)仿制藥申請(qǐng)費(fèi),也不是全歐盟統(tǒng)一的,每個(gè)國家有每個(gè)國家的標(biāo)準(zhǔn)。總的來說大概在幾千歐元到一萬多歐元。
費(fèi)用方面,歐盟的GMP現(xiàn)場(chǎng)檢查和和美國也有不一樣,在美國都是FDA自己掏錢,而歐盟GMP檢查的吃住行都是企業(yè)安排。這個(gè)是不一樣的。
7、歐盟ASMF、CEP文件簡介
做仿制藥就要原料藥,歐盟的原料監(jiān)管有兩條途徑,一條是CEP,一條是ASMF(以前叫EDMF,現(xiàn)在統(tǒng)一叫ASMF)。CEP比較簡單,歐州藥典收錄了,你就申請(qǐng)CEP。一旦CEP搞定,你在歐盟就通吃了,任何一個(gè)國家都可以去銷售和使用你的原料藥。另外一個(gè)途徑是ASMF,如果這個(gè)藥EP還沒收錄,中國藥廠做仿制藥就是報(bào)ASMF,可以單獨(dú)報(bào),也可以和制劑一起申報(bào)。
歐盟的ASMF雖然結(jié)構(gòu)和美國DMF主體相同,但是他和美國DMF最大的就不同就是ASMF在歐盟它分為公開部分和保密部分。如下所示:
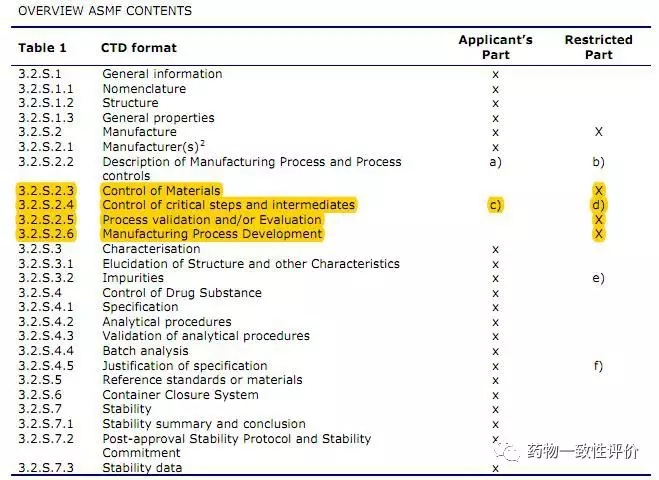
可以看到標(biāo)黃的地方,就是保密的,原料藥廠家的原料控制、生產(chǎn)工藝、過程,可以對(duì)制劑申請(qǐng)人保密。制劑申請(qǐng)人根據(jù)這個(gè)表格,可以向原料廠家索要公開部分,再把這些公開部分的資料利用到制劑的申報(bào)資料中。
在評(píng)審的時(shí)候,分為原料評(píng)審和制劑評(píng)審。制劑廠家只能收到制劑的評(píng)審意見,和原料藥的公開部分的意見。原料藥保密部分的意見只會(huì)發(fā)給原料藥廠家,制劑廠家是看不到的。這時(shí)候制劑廠家就要及時(shí)和原料廠聯(lián)系,一起配合,給官方提供的回復(fù)。
8、歐盟QP制度簡介
QP質(zhì)量受權(quán)人,在中國,大家都很熟悉,一般來說就是藥企的質(zhì)量負(fù)責(zé)人的老大,負(fù)責(zé)產(chǎn)品放行。在歐盟,這個(gè)質(zhì)量授權(quán)人和中國的質(zhì)量受權(quán)人的職責(zé)還是略有一點(diǎn)不一樣。
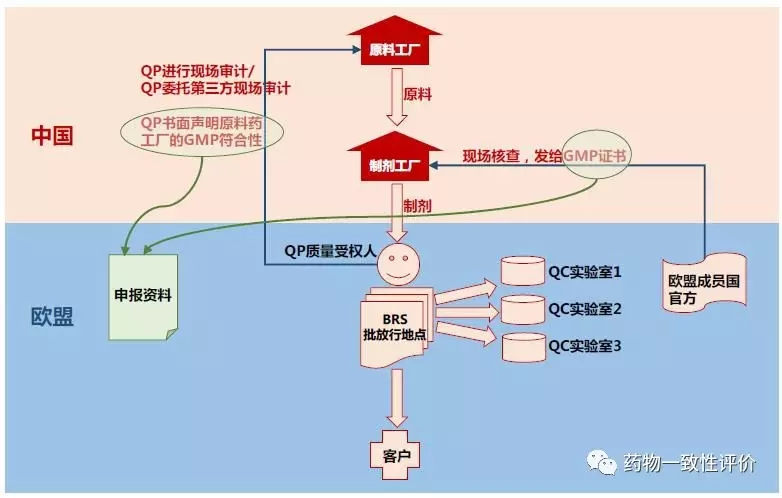
在談到歐盟質(zhì)量受權(quán)人這張圖是最形象的。以中國企業(yè)出口為例,紅色部分代表中國境內(nèi),藍(lán)色部分代表歐盟境內(nèi)。產(chǎn)品從最上面原料藥原料工廠到制劑工廠,然后運(yùn)送到歐盟。
藥品進(jìn)入歐盟,首先面對(duì)的第一關(guān)是質(zhì)量授權(quán)人,他會(huì)安排對(duì)產(chǎn)品進(jìn)行檢驗(yàn)。這個(gè)檢驗(yàn)可以在不同的實(shí)驗(yàn)室,也可以在不同的實(shí)驗(yàn)室,或者找第三方實(shí)驗(yàn)室。檢驗(yàn)完成后,匯總到批放行地點(diǎn),質(zhì)量授權(quán)人來審核這些檢結(jié)果。根據(jù)檢驗(yàn)結(jié)果來決定這些批次能不能放行。
質(zhì)量授權(quán)人難道他只負(fù)責(zé)檢驗(yàn)放行嗎?不是的。我們先看最上面原料工廠。歐盟和美國最大的不同是歐盟官方一般不會(huì)去檢查你的原料工廠的GMP符合情況。特別是你走DCP這樣的申報(bào)途徑的話,他不會(huì)去檢查。那么誰來檢查呢?就是質(zhì)量授權(quán)人
質(zhì)量授權(quán)人他只是一個(gè)有資質(zhì)的一個(gè)專業(yè)人員,他并不代表官方。他檢查完原料工廠是不會(huì)給原料工廠發(fā)GMP證書的。他會(huì)發(fā)出了一份叫QP聲明的文件,具有正常重要的法律效力。這個(gè)QP聲明是必須要再提交資料的時(shí)候要放進(jìn)申報(bào)資料的,沒有QP的聲明的申報(bào)資料是不完整的,格式審查就不會(huì)通過。
再往下看制劑工廠,制劑工廠的GMP證書是歐盟官方來檢查的,一般是某一個(gè)成員國國家安排人員來檢查,發(fā)給的GMP證書也是需要放進(jìn)申報(bào)資料。如果申報(bào)資料中沒有GMP證書也是不完整的。
從這個(gè)圖上就可以看出來,既然這兩個(gè)東西都要放進(jìn)申報(bào)資料,那歐盟在申報(bào)之前你就把GMP給過了,包括原料工廠和制劑工廠。這對(duì)于首次申報(bào)出口歐盟的廠來說,這就成了一個(gè)先有雞后有蛋的問題。那我才第一個(gè)品種申報(bào),我哪兒來的GMP證書。就需要在申報(bào)資料里面,先寫一個(gè)聲明,我先提交申報(bào)資料,我以提交申報(bào)資料來觸發(fā)歐盟官方對(duì)于我制劑工程的GMP核查。當(dāng)然,不管你是新廠還是老廠,你每提交一個(gè)品種,首先要提供原料藥工廠的QP聲明。這個(gè)是在申報(bào)之前必須要獲得,這個(gè)工作要提前去安排。
QP對(duì)原料藥工廠的GMP檢查需要在申報(bào)之前就做完,這點(diǎn)和美國還是不太一樣的。在美國,如果是一個(gè)新廠想到美國去申報(bào)仿制藥的話,一般都是先申報(bào)提交資料后,F(xiàn)DA才可能來廠里的檢查,包括原料和制劑工廠。
可能大家關(guān)注的是歐盟的QP對(duì)原料工廠的GMP審計(jì)。QO是一個(gè)人,他也不一定自己來原料藥工廠,他可以委托一個(gè)相對(duì)中立的第三方來檢查。檢查的尺度相對(duì)來說也沒有官方那么嚴(yán)。和美國最大不一樣的是,QP在提交原料藥的QP聲明的時(shí)候,原料藥廠必須要完成工藝驗(yàn)證。這和美國提交DMF所要的數(shù)據(jù)相比和內(nèi)容相比要多的多,美國DMF只要過一個(gè)CA就可以了。歐盟至少要做完工藝驗(yàn)證才能給你發(fā)這個(gè)QP說明。就這一點(diǎn)在項(xiàng)目規(guī)劃和管理上必須要考慮到,否則被卡在這里就很被動(dòng)了。
再展開來說一說,歐盟和美國申報(bào)的不一樣。這個(gè)圖上也能看出來這個(gè)藍(lán)色的指歐盟。藥品要進(jìn)入歐盟,并不是說在中國的工廠檢驗(yàn)了,拿著一個(gè)合格報(bào)告到歐盟就能直接發(fā)給客戶去使用。而是,進(jìn)歐盟以后必須經(jīng)過質(zhì)量受權(quán)人重新的完整的檢驗(yàn),開檢驗(yàn)報(bào)告才能夠放心,這點(diǎn)是和美國比較不同的。這個(gè)叫批檢驗(yàn)或者批放行,關(guān)于批檢驗(yàn)批放行的地點(diǎn),歐盟規(guī)定這個(gè)地點(diǎn)必須在歐盟境內(nèi)。
結(jié)合剛才的舉例,如果說你的主審國是德國副申國意大利和英國三個(gè)國家,對(duì)于批放行地點(diǎn),并不要求你一定要在這三個(gè)國家,你可以選擇一些相對(duì)來說經(jīng)濟(jì)沒那么發(fā)達(dá),用人成本比較低的國家,比如說匈牙利、保加利亞之類的地方。只要有正常的歐盟的GMP實(shí)驗(yàn)室資質(zhì)就行,藥品可以先發(fā)到匈牙利,檢驗(yàn)完成后,再發(fā)到英國去,就可以節(jié)約成本。
9、歐盟藥物警戒
藥物警戒,國內(nèi)習(xí)慣稱為不良反應(yīng)監(jiān)控。美國仿制藥申請(qǐng)的時(shí)候,藥物警戒在申報(bào)資料中是不需要體現(xiàn)的。而歐盟則不同,需要在在提交申請(qǐng)的時(shí)候就準(zhǔn)備好。這就意味著中國企業(yè)想去歐盟申報(bào),這塊工作必須得提前準(zhǔn)備。
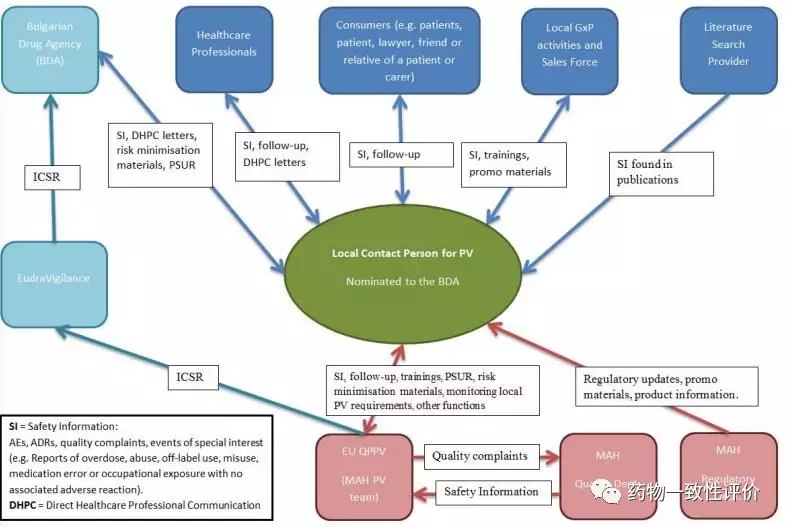
以上是比較典型的一份歐盟藥物警戒的流程圖。做很簡短的介紹。從最左邊開始看起,上面是保加利亞的藥政官方,下面是歐盟的藥物警戒的一個(gè)數(shù)據(jù)庫。那么我們先看最中間這個(gè)的當(dāng)?shù)芈?lián)系人,一般來說,這也是需要有一定的資質(zhì)的授權(quán)人。他負(fù)責(zé)收集醫(yī)生、消費(fèi)者、當(dāng)?shù)氐倪@些銷售商、文獻(xiàn)信息等等,這些信息匯總他這兒以后。他會(huì)交給最下面一行的QPPV。QPPV又需要和持有人的質(zhì)量部門進(jìn)行反饋。
QPPV除了跟工廠的質(zhì)量部門進(jìn)行溝通之外,他還需要把這些不良反應(yīng)這些信息匯總到歐盟的總的數(shù)據(jù)庫里面去。就有點(diǎn)類似我們國內(nèi)的這個(gè)不良反應(yīng)監(jiān)測(cè)體系的一個(gè)功能。
在申報(bào)資料中除了要詳細(xì)描述QPPV負(fù)責(zé)的藥物警戒體系,還需要提交對(duì)于藥物上市后監(jiān)管的風(fēng)險(xiǎn)管理計(jì)劃RMP,這和美國都是不一樣的。
10、非集中程序申報(bào)流程
下面介紹最為典型的一個(gè)仿制藥申請(qǐng)的流程,從最接近實(shí)際操作的DCP為例。
首先,你要選擇主審國和副審國,歐盟的主審國是需要提前預(yù)約的,這和美國和中國是不一樣的。我們知道,中國和美國歷史上都發(fā)生過評(píng)審積壓,歐盟是沒有出現(xiàn)過,這就和歐盟的預(yù)約制度是有關(guān)系。預(yù)約制度是指的是提提交申請(qǐng)之前需要和你的主審國提前預(yù)約檔期,否則不會(huì)受理。
例如選擇荷蘭為主審國,你就需要先去看荷蘭有沒有檔期。
http://dcp-time-slot.cbg-meb.nl/
這是典型的荷蘭官網(wǎng)的可預(yù)約表,中文可以叫預(yù)約檔期。只有給了你這個(gè)檔期了,你才能提交。從網(wǎng)上可以看出來了,荷蘭的檔期是非常的滿。他的評(píng)審組分為五個(gè),其中四個(gè)是化藥,一個(gè)是這個(gè)植物藥。化藥根據(jù)適應(yīng)癥又被分為不同的組。目前是2018年4月,看到很多組比如第二組到8月份檔期都已經(jīng)約完了。如果你這個(gè)時(shí)間存在第二組提交資料,你最快也要等到九月份才能提交。
歐盟的整體來說,為什么沒有積壓呢,因?yàn)樗o的檔期是有限的,數(shù)量也比較少,比如說荷蘭,荷蘭是一個(gè)非常典型的主審國。1個(gè)月1個(gè)組才給4個(gè)申請(qǐng)檔期。也就是說,以2018年為例,1個(gè)月給合計(jì)4個(gè)組也就給16個(gè)申請(qǐng)。一年12個(gè)月合計(jì)整個(gè)荷蘭評(píng)審200個(gè)左右。所以我們就感嘆,像荷蘭這樣的國家,為什么沒有積壓,或者整個(gè)歐盟他沒有積壓,他沒有申請(qǐng)哪兒來積壓?他一個(gè)月總共才受理16個(gè)申請(qǐng),他怎么可能積壓?而且荷蘭在歐盟還是作為主審國最多的國家。荷蘭因?yàn)楸容^忙,所以我們從這個(gè)網(wǎng)頁上可以看到,他從2019年開始,他們每個(gè)月的每一個(gè)評(píng)審組的檔期上升到五個(gè)了。
在歐盟,不是所有國家的預(yù)約檔期都像荷蘭如此公開,或者說大部分國家都是不公開的,你只能寫信去問或者申請(qǐng)。對(duì)于這個(gè)申請(qǐng)的回復(fù)的期限,不同國家都有自己的規(guī)定,快的很快一兩周就能回復(fù),慢的等兩三個(gè)月才給你回復(fù),效率還是很慢的,藥政上要提前規(guī)劃好。
第二個(gè)更重要的就是,DCP動(dòng)程序申報(bào)中,主審國和副審國的選取。主審國肯定選擇選擇比較有實(shí)力的國家。這個(gè)是HMA網(wǎng)站上公布的2017年作為主審國作DCP的國家,排名前五的是荷蘭、德國、英國、葡萄牙和瑞典,第六名的是丹麥。
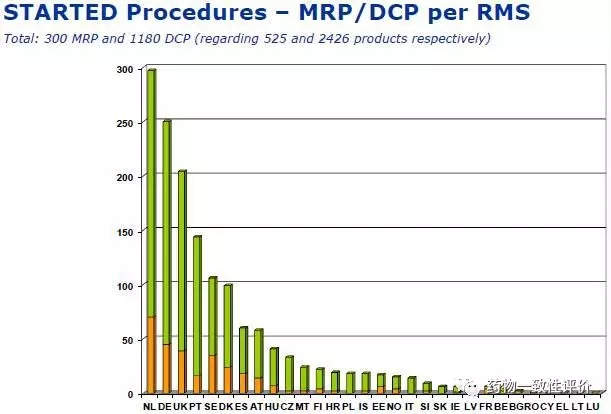
 對(duì)于中國企業(yè)來說呢,建議選擇評(píng)審數(shù)量比較多的國家最好。有人可能要問,他們的評(píng)審那么多,我們就不去排隊(duì)了吧。但是你要注意,如果你選擇評(píng)審數(shù)量比較少的國家,他很可能沒有經(jīng)驗(yàn),沒有人手,在某些CMC問題上會(huì)給你提供一些不專業(yè)的建議,讓你還沒法反駁,所以我們不建議選擇當(dāng)主審比較少的國家。
對(duì)于中國企業(yè)來說呢,建議選擇評(píng)審數(shù)量比較多的國家最好。有人可能要問,他們的評(píng)審那么多,我們就不去排隊(duì)了吧。但是你要注意,如果你選擇評(píng)審數(shù)量比較少的國家,他很可能沒有經(jīng)驗(yàn),沒有人手,在某些CMC問題上會(huì)給你提供一些不專業(yè)的建議,讓你還沒法反駁,所以我們不建議選擇當(dāng)主審比較少的國家。
副審國我們一般要根據(jù)市場(chǎng)和商業(yè)情況等選擇。如果這個(gè)地區(qū)這個(gè)藥你覺得有商業(yè)潛力商業(yè)合作伙伴比較強(qiáng)大,那你可以選擇副審國。一開始我們也介紹的主審國和副審國,中國的企業(yè)一般選擇都不會(huì)太多,一個(gè)主審國,2-5個(gè)副審國就屬于比較多,超過五個(gè)的很少。
典型的DCP評(píng)審流程
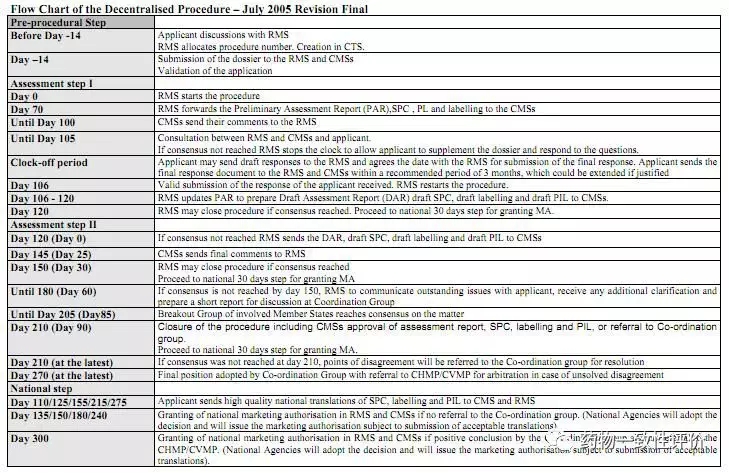
這個(gè)圖大家可能不一定看得清楚,那么在網(wǎng)上很容易檢索到這個(gè)表格,挑幾個(gè)關(guān)鍵節(jié)點(diǎn)介紹一下。評(píng)審大概可以分為三個(gè)大的階段:
第一個(gè)階段就申報(bào)正式受理之前的14天,表格上看上去-14天。其實(shí)它指的就是這個(gè)申報(bào)資料的格式審查。如果有缺陷,他會(huì)跟你聯(lián)系補(bǔ)正。一般來說,這個(gè)工作會(huì)在14天之內(nèi)就能解決。但如果你的回復(fù)慢,這個(gè)就可能延長到后面去。
評(píng)估第一階段。相當(dāng)于是正式的一個(gè)受理日期,Day0開始,并進(jìn)入到這個(gè)評(píng)審階段。70天后,主審國就要把他的評(píng)審意見發(fā)給申請(qǐng)人,這個(gè)速度還是非常快的。那么105天左右,副審國的評(píng)審意見也會(huì)發(fā)給主審過,并一起發(fā)給申請(qǐng)人。
評(píng)估第二階段。申請(qǐng)人根據(jù)105天的匯總的評(píng)審已將,進(jìn)行回復(fù),以及主審國和副審國之間進(jìn)行溝通,直至批準(zhǔn)上市。
當(dāng)然總的流程和美國還是比較接近,一個(gè)仿制藥要準(zhǔn)備12到18個(gè)月,這還算是比較順利的情況。
11、申報(bào)資料
我主要關(guān)注和美國的不同的地方。例如:
-
專家信息,包括藥學(xué)的、臨床的、非臨床的,三方面專家提供信息。這個(gè)是美國不需要而歐盟需要的。
-
第二個(gè),藥物警界系統(tǒng)的建立和風(fēng)險(xiǎn)管理計(jì)劃。
-
第三,我們前面已經(jīng)提到QP聲明。它實(shí)際上是指對(duì)原料工廠的GMP保證的一個(gè)聲明。
-
歐盟的這M1里面,也會(huì)有說明書相關(guān)的信息,但是又和美國有不同。主要體現(xiàn)在以下兩個(gè)方面:1)歐盟的說明書,它需要進(jìn)行就是可讀性測(cè)試,這個(gè)是美國不要的。在美國,你就照抄原研就行了,而在歐盟需要做可讀性測(cè)試。關(guān)于可讀性測(cè)試,一句話描述,他就是對(duì)你的說明你起草的說明書,普通的老百姓來讀會(huì)不會(huì)有困難。如果用的詞太專業(yè),或者說寫的太凌亂,普通老百姓都讀不懂,你可讀性測(cè)試就沒法通過。可讀性測(cè)試是可以交給第三方去做。他們會(huì)安排不同的人來閱讀,然后反饋,反正完了以后你去做修改。當(dāng)然了,對(duì)中國藥企進(jìn)歐盟,這個(gè)工作肯定是交給第三方做。2)歐盟和美國說明書不一樣的。歐盟包裝裝盒上面就是要去做盲文就是布萊爾盲文,在美國是沒有要求的,這個(gè)花點(diǎn)時(shí)間學(xué)一下也是非常簡單的
-
最后很大的不同就是在工藝驗(yàn)證信息上,美國一般不需要在ANDA提交時(shí)完成工藝驗(yàn)證,只需要展示批(穩(wěn)定性批)三批即可。而歐盟的要求則多很多,有很多類型的申報(bào)都需要在申報(bào)提交時(shí)就完成工藝驗(yàn)證,具體可以參考?xì)W盟指南:21 November 2016 Guideline on process validation for finishedproducts - information and data to be provided in regulatory submissions.