
1月12日,CDE許嘉齊主任主持召開了“抗PD-1/PD-L1單抗申報資料要求專題研討會”,參會人員包括正在開展PD-1/PD-L1藥物臨床試驗的部分企業代表、臨床KOL以及CDE的腫瘤藥審評專家。這次會議的內容主要是討論PD-1/PD-L1研發中的問題和申報資料的準備,以引導創新藥物的研發,加快臨床急需藥品的上市。
2月8日,CDE對外公布了此次會議形成的共識——《抗PD-1/PD-L1單抗品種申報上市的資料數據基本要求 》,再次引起行業人員的強烈關注。對上述共識的要點概述如下:
-
允許企業基于以ORR(客觀緩解率)為主要終點的單臂臨床試驗的結果向CDE提出有條件上市申請。
-
允許企業以滾動申請的形式,分階段提交臨床數據。對不同階段提交資料的基本要求如下:

-
企業在提交上市申請前,須先提出pre-NDA會議申請。CDE根據品種的具體情況決定是否召開pre-NDA會議以及召開會議的形式。符合上述申報資料要求的企業可按程序提交上市申請,并同時提出優先審評申請。
對于這一共識文件出臺的意義,有業內人士解讀為“國產PD-1的競跑將在新的游戲規則下重新啟動,1個多月前成功提交首個國產PD-1上市申請的信達生物或將重返起跑線,再次參加新一輪申報競賽”。
醫藥魔方為此連線信達生物董事長俞德超博士,尋求他對CDE這份文件的看法。俞德超說:“PD-1/PD-L1是腫瘤免疫治療最熱門的藥物,國內在研廠家眾多,行業關注度極高。在首個國產PD-1申請上市后,CDE迅速召開專項研討會明確PD1類藥物的申報資料要求和申報規則,一方面說明中國藥監機構對PD-1這樣一種前沿藥物的監管標準還在不斷探索調整;另一方面也可以看到中國藥監機構對此持高度嚴謹的態度。對于CDE發布的最新申報要求,我們不感到意外,我們會一如既往地支持CDE一切有利于藥物創新的改革。”
在FDA的藥品監管體系中,對于一些針對臨床需求未得到滿足的嚴重疾病藥物,為了縮短研發時間加快上市以滿足患者用藥需求,FDA在2012年開設了加速批準(Accerelated approval)通道,允許企業基于替代終點提交上市申請。所謂替代終點,是指可以預測臨床獲益的實驗室指標、影像結果等。比如腫瘤藥的臨床試驗硬指標是生存期(OS)、無進展生存期(PFS),但獲取時間較長,FDA就可以基于客觀緩解率(ORR,比如腫瘤大小縮小30%以上)的替代終點來有條件批準某個藥物上市。
CFDA此前未明確出臺過關于滾動申請和有條件批準的監管政策。隨著國內企業研發實力的提升,跟進PD-1,CAR-T等國際前沿療法的速度越來越快,這對中國的創新藥監管提出了更高的要求。CFDA目前也在全面向FDA的先進管理制度學習靠攏。
PD-1單抗是腫瘤免疫治療最主流的藥物,自2014年Opdivo首次在日本上市以來,全球共有5個PD-1/PD-L1藥物獲得FDA批準上市,2017年,這5個藥物的全球銷售額累計近100億美元。
PD-1/PD-L1藥物全球銷售額(億美元)
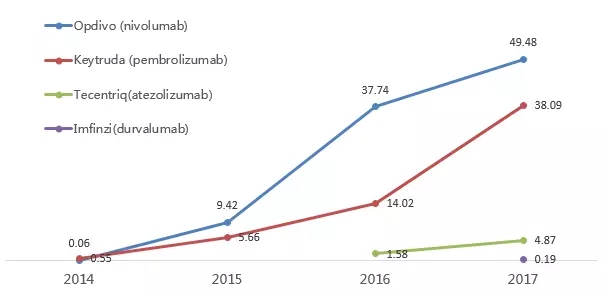
注:1)Tecentriq銷售額單位為億瑞士法郎
目前國內尚無PD-1/PD-L1藥物獲批上市,Opdivo在2017年11月率先申報上市并獲得了CDE的優先審評,2018上半年大概率獲得CFDA批準上市。國內已經申注冊申報的PD-1/PD-L1品種則多達19個,少部分企業已推進到了III期階段。